- To look back billions of years ago.
- To test the evolutionary hypothesis, "whether sequences share a common ancestor"
- Structure modeling (Homology modeling): Identify one more known protein structures likely to resemble the structure of the query sequence through comparative analysis.
Sequences that are usually aligned
- Nucleic acid sequences/DNA [4 bases - A,C,G,T]
- Protein amino acid sequences [20 Amino acids]
Techniques used to align them
- Identity - dot plots
- Scoring matrices: This also has applications in,
- Models for evolution i.e. How sequences evolved?
- Dynamic Programming algorithms
- Evaluating the significance of pairwise alignment (?)
Scoring or Comparison Matrices
In order to align the sequences the algorithm has to know what it is looking for and how it can evaluate the worth of what it finds using 'comparison matrices'. Comparison matrices define a score for every possible match possibility - a measure to quantify how well the computational alignment is doing in the context of 'sequence alignment'.
Changes can occur in the sequence by way of:
- Substitution
- Insertion
- Deletions
- Transposition - Due to DNA breakage and re-joining
- Inversion
- Recombination
- Duplications
We can always determine an optimal alignment between any two sequences. The importance lies in determining the significance of this alignment. And, for that we need to look at the structural similarities. e.g. homology modeling in protein.
Two important types of alignment
- Local - Alignment between s1 and s2 (end to end)
- Global
- Alignment between substring of s1 and s2
- Looks for local regions of similarity
- Needleman Wunch algorithm (guarantees optimal alignment)
Algorithms for both local and global alignments use Dynamic Programming techniques. Dynamic programming techniques divides the problem into identical smaller sub-problems. The best solution to one sub problem is independent from the best solution to the other sub-problem.
Selecting scoring matrices and parameters
Selecting scoring matrices and parameters is one problem
Adjust scoring for gaps
- Penalty for gaps (insertions and deletions)
- There is no theory for penalizing gaps
- Based on trial and error.
- Different scoring matrices differ in gap penalty as well.
Evaluating statistical significance
Based on Homology modeling?
Dot Plots
- Diagonals easily allow to visualize the regions of similarity.
- Visually conveys a lot of information. e.g. regions of identity, duplication
- Really high noise in case of nucleotides. just 4 bases
- To reduce the noise: Look for identity over a window (window size and minimum score in that window).
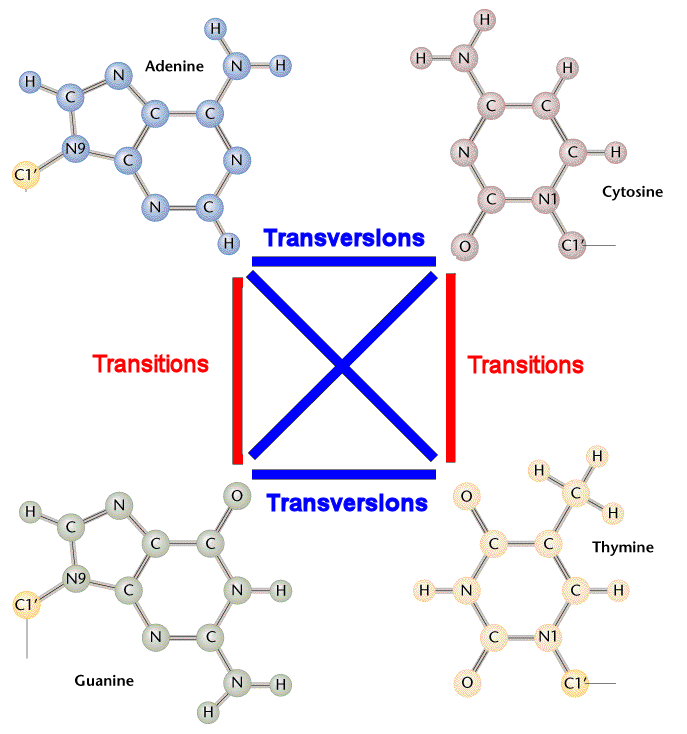
Transitions Vs Transversions
Do all nucleotide changes occur with equal frequency? There are twice as many transversions than transitions. (see diagram)
Figure 1. Transition Vs Transversion mutations (source: Steve M. Carr)
Frequency of transitions is much higher than that of transitions in nature. Thus, transitions and transversions are penalized differently.
Are all positions equally likely to change?
Including indels
In this case we don't know whether there was a transversion or transition.
Terminal gaps are treated differently from internal gaps.
Note: Optimal global and local alignments can be computed in O(|s1|.|s2|) run-time and O(|s1| + |s2|) space complexity.
Pairwise Sequence alignment
- Optimal, polynomial time algorithms known.
Multiple Sequence alignment
- NP hard. Approximate algorithms based on pairwise sequence alignment
References and useful links
1. Dynamic programming and sequence alignment
{kind=link}
2. Pairwise sequence alignment - Its all about us!
3. Homology modeling
4. Class notes and lectures. - Prof. Jung Choi, BIOL 8803
Nice post, Pushkar!! :)
ReplyDelete